Publié le
Lecture 18 mins
Hypertension pulmonaire : classification et outils diagnostiques
O. SITBON, Service de pneumologie et soins intensifs, hôpital de Bicêtre, Inserm UMR-S 999, université Paris-Sud, Le Kremlin-Bicêtre
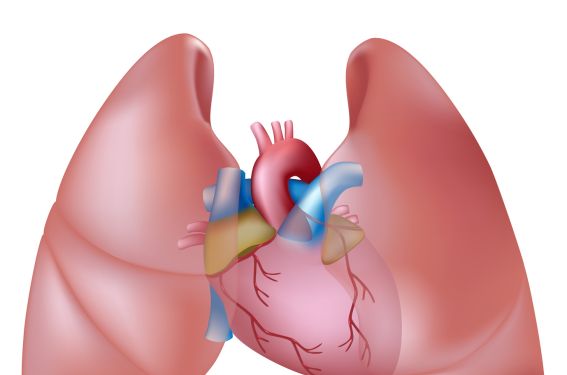
L’hypertension pulmonaire (HTP) est caractérisée par l’augmentation progressive des résistances artérielles pulmonaires aboutissant à une insuffisance cardiaque droite et au décès. Au cours des dix dernières années, les progrès réalisés dans l’épidémiologie et la compréhension des mécanismes physiopathologiques des HTP ont permis une classification clinique plus précise, indispensable au clinicien pour prendre en charge cette affection.
L’ HTAP est une forme particulière d’HTP caractérisée par la prolifération intense de la paroi des petites artères pulmonaires. Il s’agit d’une maladie rare pour laquelle des progrès très importants ont été réalisés au cours des quinze années, avec la mise au point de plusieurs molécules innovantes...
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :



