Publié le
Lecture 10 mins
Le spectre des pneumopathies interstitielles diffuses chroniques liées au tabac
Robert BARBIER*, Guillaume GRANIER**, Irina LATU*** - *Centre de pneumologie, Clinique Rhône Durance, Avignon ; **Anatomopathologie, Centre hospitalier, Avignon ; ***Pneumologie, Centre hospitalier, Avignon
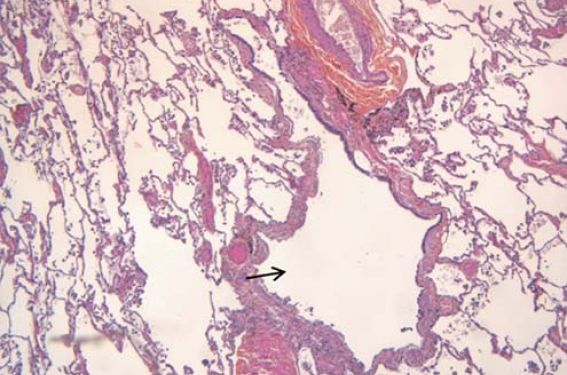
Les pneumopathies interstitielles diffuses liées au tabac constituent un éventail complexe de pathologies qui reste à ce jour encore mal cerné. Les raisons d’une relative confusion sont multiples : un certain nombre de ces pneumopathies sont rares, la terminologie est volontiers empruntée aux anatomopathologistes et il a fallu en retrouver la correspondance radio-clinique, de nombreux acronymes en langue anglaise ont vu le jour et il a fallu créer des équivalents en français (tableau). En outre, il existe des chevauchements entre certaines de ces entités. Le diagnostic anatomopathologique formel nécessite de larges biopsies chirurgicales qui ne sont que rarement exécutées dans la pratique clinique et le diagnostic est donc souvent présomptif. Pour finir, de nouvelles entités sont décrites par les pathologistes comme le « Smoking Related Interstitial Fibrosis » sans que sa pertinence clinique ait été clairement reconnue.
Les différentes pneumopathies interstitielles diffuses chroniques liées au tabac La dernière classification des pneumopathies interstitielles diffuses idiopathiques ATS/ERS date de 2013 (1). Entre autres précisions par rapport à l’édition de 2002, elle introduit la notion de pneumopathies...
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :



